top of page
This site was designed with the
.com
website builder. Create your website today.
Start Now
CASUAL CHEMISTRY
Home
Blog
More
Use tab to navigate through the menu items.
Log In
CASUAL CHEMISTRY
Presenting topics in Chemistry in an accessible and grounded way
Home: Welcome
Play Video
Play Video
20:40
Casual Chemistry
A Retrosynthetic Analysis of Aspidophytine
A proposal for a disconnection approach to analysing this alkaloid natural product. The retrosynthesis is driven by key functional group relationships and minimising competition between electrophiles and nucleophiles. This retrosynthetic analysis identifies sensible starting materials as an indole and a cyclopentenone (ketone functionalised cyclopentene). This discussion was inspired by the Corey synthesis of apspidophytine: J. Am. Chem. Soc. 1999, 121, 28, 6771–6772 https://pubs.acs.org/doi/10.1021/ja9915201 Links to other mentioned videos: - CBS reduction: https://youtu.be/rv-dZ2qyAzM - Indole synthesis: https://youtu.be/v2lSd253nwU
Play Video
Play Video
00:55
Casual Chemistry
Prevost Reaction in Organic Chemistry #ochem #chemistry #organicchemistry
Prevost dihydroxylation reaction is selective for converting an alkene to an anti diol, using an intermediate iodonium ion, SN2 substitution and neighbouring group participation.
Play Video
Play Video
01:01
Casual Chemistry
Anti Aldol Reactions - Lactate Auxiliaries #chemistry #organicchemistry #ochem #chemistryeducation
Play Video
Play Video
22:07
Casual Chemistry
Solving Anti Aldol Reaction Issues
Using lactic acid derived auxiliary chemistry to perform highly diastereoselective anti aldol reactions (lactate aldols). A full explanation evolving a Zimmerman-Traxler Transition State into a chelated boat-like transition state where the configuration of an alpha (prime) stereocentre on the boron enolate can induce stereochemical control on the product. The reaction relies on a stabilising, non-classical hydrogen bond with the formyl hydrogen atom of the activated carbonyl group. Links to other videos referenced --------------------------------------------------- Introduction Part 1: https://youtu.be/XZ30Fup3xyA Introduction Part 2: https://youtu.be/qFe5T7WLHDY Felkin-Anh Model: https://youtu.be/JvF5NQ54-z4 A simple boron-mediated aldol reaction: https://youtu.be/b9KWPWeVkZg A retrosynthesis using auxiliary chemistry https://youtu.be/zlclJzdrtBI References from video ------------------------------------ Studies on Lactate Aldol Reactions: Tetrahedron Lett. 1994, 35, 9083-9086 https://doi.org/10.1016/0040-4039(94)88434-X Tetrahedron Lett. 1994, 35, 48, 9087-9090 https://doi.org/10.1016/0040-4039(94)88435-8 J. Org. Chem. 1992, 57, 19, 5173–5177 https://doi.org/10.1021/jo00045a033
Play Video
Play Video
00:15
Casual Chemistry
Barton-McCombie Deoxygenation #organicchemistry #chemistry #orgo
For removing a hydroxyl group using controlled radical chemistry. The overall transformation reduces an alcohol to an alkyl group.
Play Video
Play Video
00:15
Casual Chemistry
Ellman Auxiliaries for Amine Synthesis #chemistry #organicchemistry
For asymmetric synthesis of primary amines. A stereocentre is formed by direct addition of a Grignard nucleophile to a sulfinimide (sulphinimide) with a stereogenic sulfur centre. The sulfinimide is formed by condensation of the enantiomerically enriched sulfoxide (sulphoxide) with a carbonyl, such as an aldehyde or ketone. The transition state is thought to be a chelated Zimmerman-Traxler type six-membered ring.
Play Video
Play Video
00:16
Casual Chemistry
Eschenmoser Fragmentation #chemistry #organicchemistry
Eschenmoser fragmentation of an epoxy hydrazine. The epoxide has likely formed by conjugate addition to an alpha,beta-unsaturated ketone. The reaction is driven by base here but the fragmentation can also be started with acid. After releasing ring strain in the epoxide, a further fragmentation forms a new carbonyl and allows two really good leaving groups to leave - nitrogen gas and the tosyl anion. In this process an alkyne is formed with a triple carbon-carbon bond.
Play Video
Play Video
00:16
Casual Chemistry
Payne Rearrangement #chemistry #organicchemistry
Mechanism for the isomerisation of epoxy alcohols to the terminal epoxide. It is likely that the initial epoxide was generated by oxidation of an allylic alcohol - perhaps by asymmetric catalysis using a Sharpless Asymmetric Epoxidation reaction. The terminal epoxide is favoured as it has less steric strain from eclipsing interactions.
Play Video
Play Video
00:15
Casual Chemistry
Pummerer Rearrangement #chemistry #organicchemistry
Organic chemistry mechanism of a sulfoxide (sulphoxide) reacting with acetic anhydride to give a thiol-acetate via elimination
Play Video
Play Video
00:16
Casual Chemistry
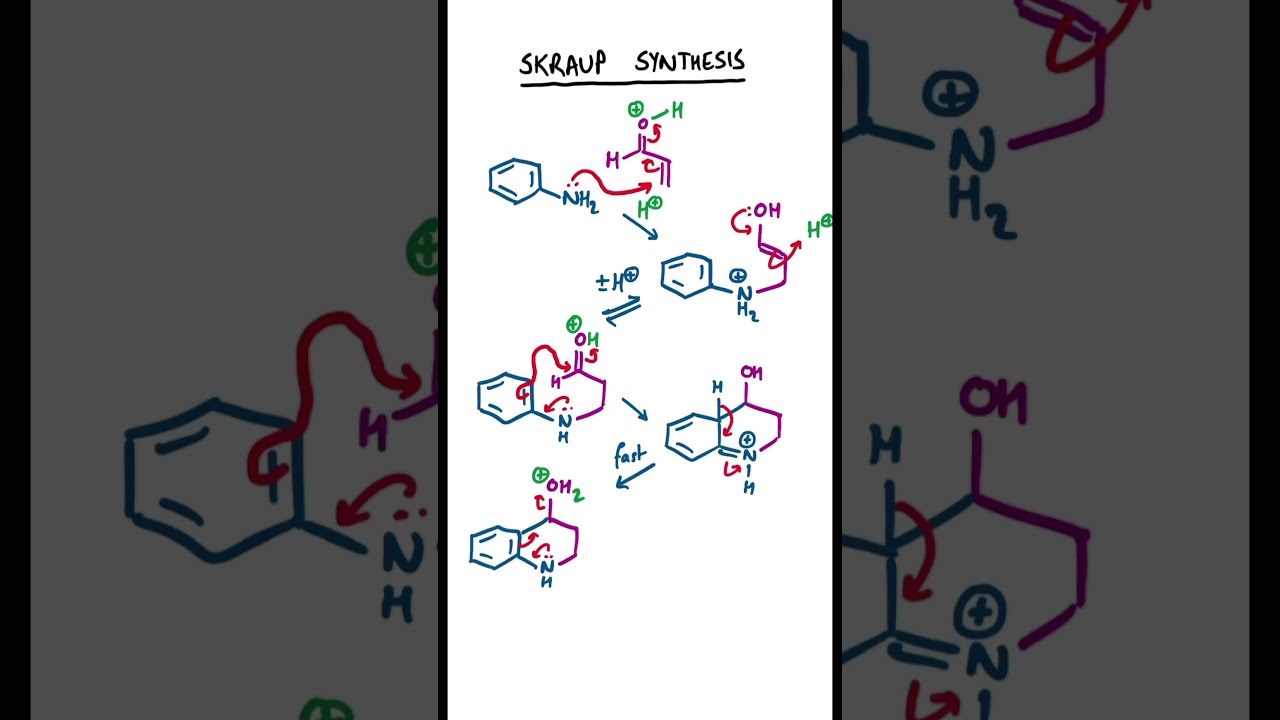
Skraup Synthesis #chemistry #organicchemistry
Synthesis of quinoline. Conjugate addition of aniline to the 1,4 position of acrolein. Mediated by acid. Cyclisation by electrophilic aromatic substitution on to an activated carbonyl group. After rearomatisation, water is eliminated likely by an E1 mechanism. The intermediate is then oxidised by a separate oxidising agent such as nitrobenzene.
Play Video
Play Video
00:16
Casual Chemistry
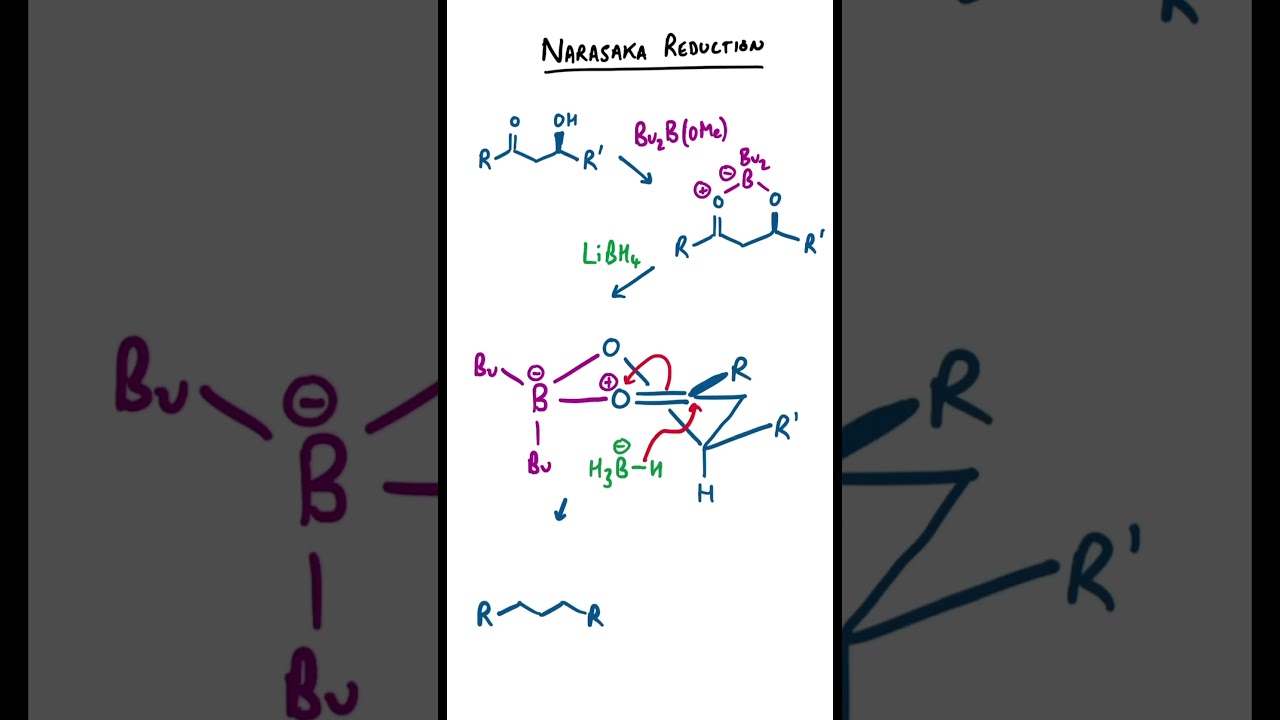
Narasaka Reduction Mechanism #chemistry #organicchemistry
Narasaka reduction mechanism where a beta hydroxy-ketone is chelated first with a boron based bidentate Lewis acid. The chelated structure prefers to adopt a half-chair six-membered ring conformation. Attack of an external borohydride nucleophile is from the face of the carbonyl that leads to a chair-like transition state (rather than a twist boat) to the 1,3-syn diol product. The reaction is very diastereoselective.
Play Video
Play Video
00:16
Casual Chemistry
Pinacol Rearrangement #chemistry #organicchemistry
Reaction mechanism for the Pinacol Rearrangement in Organic Chemistry and Synthesis. A reductive coupling using magnesium metal followed by an acid mediated migration to give a spiro centre.
Play Video
Play Video
00:15
Casual Chemistry
CBS Reduction as Asymmetric Catalysis #chemistry #organicchemistry #ochem
Chiral sodium borohydride for reduction of ketones in organic chemistry. The proline based species can be used like this as a type of asymmetric catalysis to form one enantiomer of product in high enantiomeric excess (ee).
Play Video
Play Video
00:16
Casual Chemistry
Sharpless Asymmetric Epoxidation #chemistry #organicchemistry #ochem
Named reaction in Organic Chemistry. Mechanism in longer video.
Play Video
Play Video
00:16
Casual Chemistry
Evans Auxiliaries in Aldol Reaction Chemistry
Chiral auxiliary in Organic Chemistry #chemistry #organichemistry
Play Video
Play Video
29:06
Casual Chemistry
Evans Auxiliaries and a Friend for Aldol Reactions
Using chiral auxiliaries for asymmetric synthesis - diastereoselective aldol reactions with chelating enolates and cyclic transition states. The transition states can be constructed using the Zimmerman-Traxler model. Motivation in my previous video: https://youtu.be/XZ30Fup3xyA Following selective enolisation, either by hard enolisation with strong bases such as LDA or by soft enolisation methods using a Lewis acid and a weak base, a stereodefined enolate can react with aldehydes in an aldol reaction. Both the nucleophile and electrophile are prochiral and so we form two new stereocentres from this reaction. The reaction is diastereoselective. The diastereoselectivity can be increased by using chelation into a six-membered ring transition state, in which big groups prefer to go equatorial. A chiral auxiliary can be used to select further for a particular combination of stereocentres that can be formed in this reaction. The chiral auxiliary adds another level of diastereoselectivity and the Evans auxiliary can lead to very high d.r. (diastereomeric ratio). When the aldol reaction is complete, the chiral auxiliary can be cleaved carefully, separate by chromatograhpy, and recycled. Another type of chiral auxiliary is the lactate auxiliary which uses a stereocentre derived from lactic acid. Both enantiomers of lactic acid are available in the chiral pool. There are similar types of diastereoselectivity observed with these chiral auxiliaries and this topic will be expanded upon in my next video. REFERENCES Evans Aldol Reaction: J. Am. Chem. Soc. 1981, 103, 8, 2127–2129 https://doi.org/10.1021/ja00398a058 Lactate Aldol Auxiliaries: Tet.Letters Vol. 35. No. 48, pp. 9083-9086. 1994 https://doi.org/10.1016/0040-4039(94)88434-X FURTHER DETAIL Felkin-Anh Model: https://youtu.be/JvF5NQ54-z4 Boron aldol reaction: https://youtu.be/b9KWPWeVkZg Another example of use of chiral auxiliaries: https://youtu.be/zlclJzdrtBI #chemistry #organicchemistry #education
Play Video
Play Video
00:34
Casual Chemistry
Diels-Alder Reaction
Play Video
Play Video
00:32
Casual Chemistry
Appel Reaction Mechanism in Organic Chemistry
Play Video
Play Video
17:08
Casual Chemistry
Making a Synthesis Stereoselective
Introduction to stereoselective synthesis via retrosynthetic analysis. How to change reagents without changing disconnections in a retrosynthesis to introduce diastereoselectivity and enantioselectivity. Discussion based on this literature synthesis: https://doi.org/10.1002/anie.201310164 Boron Enolates and Aldol Reactions: https://youtu.be/b9KWPWeVkZg Ultimate Guide to the Felkin-Anh Model: https://youtu.be/JvF5NQ54-z4 #chemistry #education #organicchemistry
Play Video
Play Video
16:27
Casual Chemistry
Elemental Metals in Retrosynthetic Analysis
A disconnection approach to a retrosynthesis of this organic molecule. The different ring systems can be made by classical named reactions involving metals in the forward synthesis. The 1,4-cyclohexadiene is clue for using a Birch reduction disconnection back to the benzene ring in the retrosynthesis. A mixture of sodium metal dissolved in liquid ammonia makes a solution of solvated electrons that act as a powerful reducing agent. Solvated electrons are transferred into the benzene pi system (pi cloud) to give a conjugated carbanion. This carbanion can be protonated by an external source of H+ such as an alcohol. The video includes a discussion of the reaction mechanism and the effect of both electron-withdrawing and electron-donating substituents on the regioselectivity for the Birch reduction. The cyclopropane can be synthesised by Simmons-Smith reaction. The reagent for the Simmons-Smith cyclopropanation is a carbenoid formed by mixing zinc metal with diiodomethane. In this retrosynthetic analysis, the substrate for cyclopropanation is a chiral allylic alcohol. Due to restricted rotation, high levels of diastereoselectivity for cyclopropanation should be observed as the Z geometry of alkene next to the chiral centre leads to just one low energy conformation (energy minimum for this conformer). The Simmons-Smith zinc carbenoid reagent will coordinate to the hydroxyl group on the stereocentre and direct reaction to the same face. The remainder of the retrosynthesis shows that the key intermediates can be made using simple disconnections and redox steps back to simple benzene ring systems. 1,2-diX difunctional patterns are common here which can nudge towards the use of epoxides in a forward synthesis, although other synthetic pathways are possible. #chemistry #organicchemistry #education
Load More
Home: Videos
bottom of page